


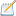

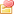

孟德尔随机化-去除连锁平衡bug解决不了呜呜
0

###################################去除连锁不平衡##################
读取暴露数据
exposure_data=read_exposure_data(filename="exposure_data.csv",
sep = ",",
snp_col = "rsID",
beta_col = "beta",
se_col = "SE",
pval_col = "P.weightedSumZ",
effect_allele_col="eff.allele",
other_allele_col = "ref.allele",
phenotype_col = "bac",
samplesize_col = "N",
chr_col="chr", pos_col = "bp",
clump=FALSE)
head(exposure_data)
dim(exposure_data)
[1] 14575 15
去除连锁不平衡的SNP
exposure_dat_clumped=clump_data(exposure_data, clump_kb=10000, clump_r2=0.001)
dim(exposure_dat_clumped)
[1] 0 15
为什么我在运行这一步之后变为0了,我用的肠道菌群数据,别人运行完也不是这样的哇.....


所有回答(1)
0
在进行孟德尔随机化分析时,去除连锁不平衡(LD)的过程正如您所描述的,如果在 clump_data 函数运行后数据的维度变为 (0, 15),这意味着没有 SNP 被保留下来。您提到使用的是肠道菌群的数据,而其他人运行时却没有此问题。这可能与以下一些原因有关:
可能的原因及解决方案:
-
输入数据中的SNP是否有重复:
- 检查
exposure_data中的 SNP 列(即snp_col)是否有重复值。clump_data函数通常依赖于 SNP 的唯一性。如果有重复的 SNP,它们可能被一并删除。 - 可以通过以下代码检查重复:
duplicate_snps <- exposure_data[duplicated(exposure_data$snp_col), ]
- 检查
-
LD 相关性:
- 您设置的
clump_r2(0.001)和clump_kb(10000)可能太严格,特别是基因组中 SNP 的 LD 结构可能导致您的数据集中没有 SNP 能够满足这个要求。尝试放宽这两个参数:exposure_dat_clumped = clump_data(exposure_data, clump_kb=10000, clump_r2=0.1) - 观察是否会保留更多 SNP。
- 您设置的
-
数据预处理问题:
- 确保
exposure_data中的 SNP 与 LD 参考面板(如 1000 Genomes 或者其他特征的基因组数据)相匹配。如果您没有提供 LD 参考,那么clump_data将无法执行。 - 确保数据类型和列名准确无误。特别注意
snp_col、beta_col、se_col等列中的数据和列名是否正确。
- 确保
-
数据完整性:
- 检查
exposure_data中的缺失值和无效数据。任何 NA 值都可能影响 clumping 过程。 - 尝试运行该行代码后加上一个检查,看是否存在 NA 值:
sum(is.na(exposure_data))
- 检查
-
兼容性问题:
- 有时您使用的 R 包版本可能会导致问题。确保
TwoSampleMR以及相关的依赖包是最新的,并与您的 R 版本兼容。
- 有时您使用的 R 包版本可能会导致问题。确保
-
运行示例代码:
- 运行一些基础的示例数据,确认
clump_data能正常运行以验证其基本功能:example_data = data.frame( rsID = c("rs1", "rs2", "rs3", "rs4"), beta = c(0.1, -0.2, 0.05, 0.3), SE = c(0.01, 0.01, 0.02, 0.03), P.weightedSumZ = c(0.05, 0.01, 0.15, 0.07), eff.allele = c("A", "G", "C", "T"), ref.allele = c("T", "A", "G", "C"), bac = c("phenotype1", "phenotype2", "phenotype1", "phenotype2"), N = c(5000, 5000, 5000, 5000), chr = c(1, 1, 2, 2), bp = c(1000, 2000, 3000, 4000) ) example_data_clumped = clump_data(example_data, clump_kb=10000, clump_r2=0.001)
- 运行一些基础的示例数据,确认
总结
请逐步验证这些方面,并优化您的参数设置。如果问题依旧,请考虑用更简化的数据,逐步检查每一步的输出,以找到问题的确切位置。如果您仍然无法解决该问题,请分享更多详细的数据样本或背景信息,以便进一步帮助。
你是不是用chatgpt回复的
